Alfa-mannosidose is een zeldzame lysosomale stapelingsziekte die zich meestal in de vroege kindertijd manifesteert.1 Het is een progressieve, uiterst heterogene ziekte die moeilijk te herkennen is en de diagnose wordt gewoonlijk pas gesteld na verwijzingen naar meerdere specialisten.1
Symptomen en rode vlaggen
Multidisciplinaire aanpak en vroege diagnose bij Alfa-mannosidose
Patiënten kunnen zich presenteren met een hele reeks klinische afwijkingen: de meeste kinderen lijken bij de geboorte normaal, maar klinische manifestaties beginnen al op erg jonge leeftijd, gevolgd door progressie van de klinische symptomen.2,3
De zeldzame en uiterst heterogene aard van alfa-mannosidose bemoeilijkt de diagnose, waardoor deze vaak laat wordt gesteld of over het hoofd wordt gezien. Dit leidt ertoe dat de ziekte vaak niet op tijd wordt gediagnosticeerd, en de lange-termijnprognose voor onbehandelde alfa-mannosidose is slecht.1
Om de patiënt een zo gepersonaliseerd mogelijke behandeling te bieden, gericht op het verbeteren van hun toestand of het remmen van ziekteprogressie, is een multidisciplinaire aanpak van groot belang.1
Een vroege diagnose, samen met de multidisciplinaire aanpak blijft het beste middel om snel met een behandeling te starten en de progressie van symptomen te beperken.1,3
Vroege diagnose van alfa-mannosidose is ook belangrijk om genetisch advies te geven, want er is 25% kans op herhaling van de ziekte (autosomaal recessieve ziekte) en ouders kunnen prenataal of pre-implantatie diagnostische procedures doorlopen.4
"Red flags" herkennen om de diagnose Alfa-mannosidose vroeg te stellen
Sommige klinische kenmerken van alfa-mannosidose zijn:2,5,6
- gehoorverlies;
- afwijkingen van het skelet;
- cognitieve stoornissen;
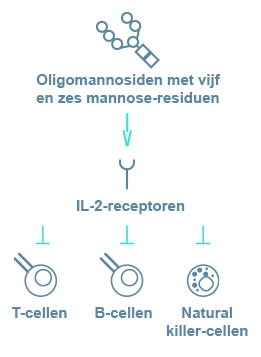
- immuundeficiëntie;
- herhaaldelijke infecties.